Pharmacotherapeutic Principles
of Neurological and Psychiatric Disorders
John A. Schetz
Summary
The pharmacotherapeutic management of neurological and psychiatric disorders relies
primarily on the modulation of central nervous system (CNS) neurotransmission with drugs
that intervene at chemical synapses. The receptors, transporters, and enzymes for the dopaminergic, serotonergic, and noradrenergic systems are the most common neuropsychiatric
drug targets, because these neurotransmitter systems play a central role in the regulation of
a range of cognitive and motor behaviors.
The key to understanding or anticipating the
clinical profile (dose–effect) of a particular drug is to have an appreciation for both its
pharmacodynamic and pharmacokinetic properties.
Key Words: Psychiatric; neuroscience; pharmacology; pharmacodynamics; pharmacokinetics; synapse; G protein-coupled receptors; dose–effect; theory; disorder; neurotransmission.
1. INTRODUCTION
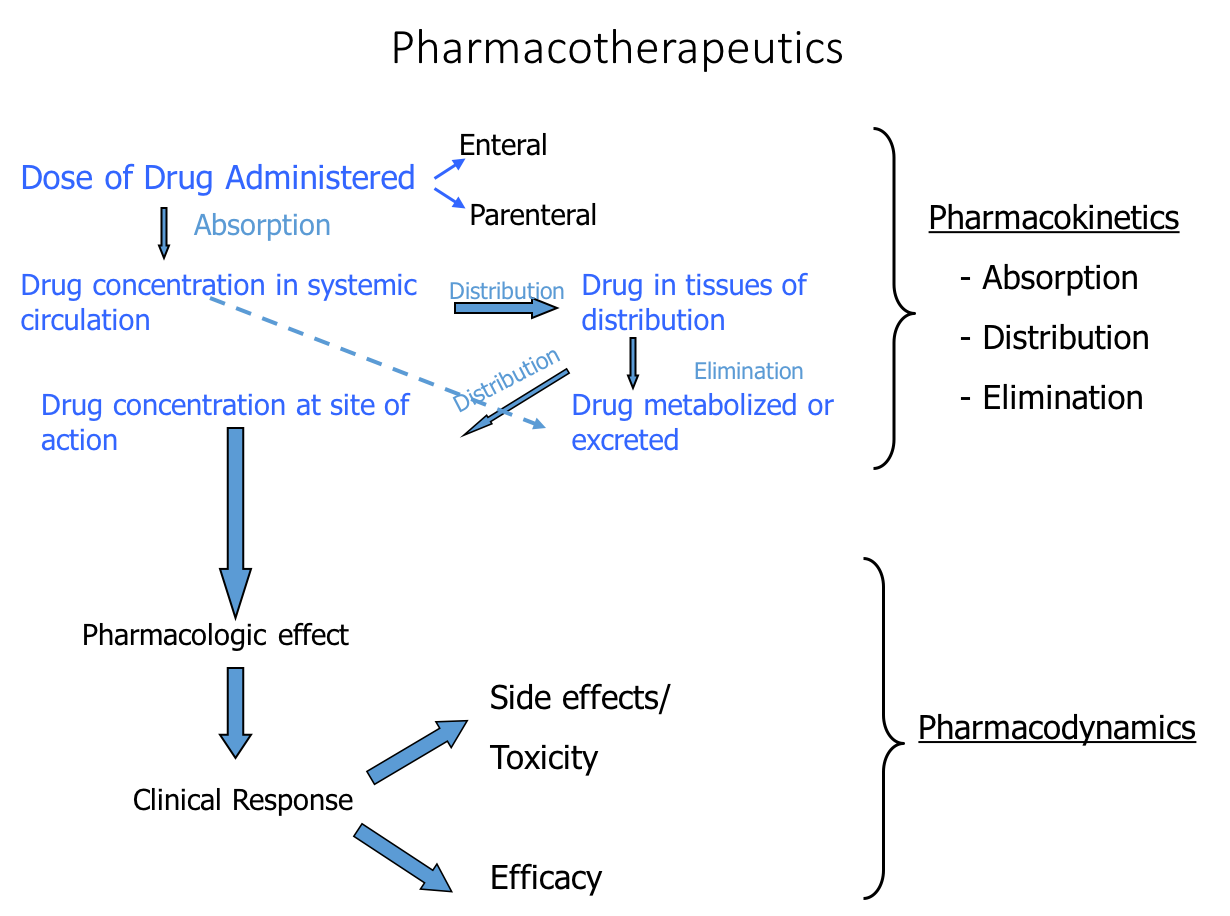
The appropriate, effective, and safe utilization of drugs in the treatment of disease requires a basic understanding of the dose–effect relationships of medications.
Relating dose to effect requires a combined appreciation of pharmacodynamic
concentration–effect relationships, or what drugs do to the body, and of pharmacokinetic dose–concentration relationships, or what the body does to or with drugs.
For this reason, considerable attention is devoted to both the pharmacodynamic and
pharmacokinetic aspects of drug therapies.
The aim of this chapter is to provide a
theoretical rationale that is necessary for appropriately interpreting the results of
basic and clinical neuropharmacology studies and for understanding many of the
drug treatment strategies commonly encountered in clinical neurology and
psychiatry.
Approximately one-fourth of all drugs prescribed worldwide exert their therapeutic actions on CNS targets. Of the top five selling drugs in this category, three
are antidepressants and two are atypical antipsychotics
(1). The relative success of
pharmacological intervention is highlighted further when one considers that these
30 Schetz
drugs are treating an estimated 7–15% of the population who suffer from one or
more of the neurological or psychiatric disorders discussed in this book. Currently,
the biogenic amine neurotransmitter systems, and in particular dopaminergic,
serotonergic, and noradrenergic receptors, transporters, and metabolic enzymes,
cover the vast majority of neuropsychiatric drug targets. The reason for this is that
the biogenic amine systems are key modulators of neuronal excitability, and the
molecular components of these systems are located at chemical synapses, which
are sites that are accessible to intervention by drugs.
2. THE CHEMICAL SYNAPSE
AS THE MAIN SITE OF DRUG INTERVENTION
Therapeutic approaches to modulating neuronal excitability at chemical synapses
can be categorized as presynaptic and postsynaptic.
Presynaptic strategies involve
altering the levels of neurotransmitter in the synaptic cleft. This can be achieved by
changing the amount of endogenous neurotransmitter released or available for
release into the synaptic cleft, or by altering the amount of neurotransmitter taken
back up (reuptake) into the presynaptic terminal.
The dopaminergic synapse can be
used as a specific example to illustrate these points (Fig. 1).
For example, monoamine oxidase B inhibitors, such as selegiline, block dopamine (DA) degradation,
which makes more DA available for release. Inhibitors of DA synthesis, such as
α-methylparatyrosine, reduce the amount of DA available for release. Drugs like
reserpine and tetrabenazine decrease vesicular-mediated release by blocking
vesicular monoamine transporters, which prevents the storage of neurotransmitter
into synaptic vesicles.
Inhibitors of plasmalemmal DA transporters, such as
buproprion or cocaine, block the reuptake of DA from the synapse, and thereby,
keep levels of DA in the synaptic cleft high. Certain drugs, like the psychostimulant
amphetamine, cause nonvesicular DA release by running the DA transporte
Pharmacotherapeutic Principles 31
31
Fig. 1.
32 Schetz
agonists, such as DA or the antiparkinsonian drug pergolide, directly activate DA
receptors, whereas neuroleptic drugs like thioridazine and haloperidol block DA
receptor activation.
3. CLASSIFICATION OF DRUGS ON THE BASIS
OF THE RESPONSES PRODUCE ON THEIR RECEPTORS
When a drug reversibly binds to the orthosteric (primary) site on its receptor one
of four outcomes are to be expected: the receptor becomes activated, the receptor
becomes partly activated, the receptor becomes inactivated, or the receptor is unable
to be activated. Consequently,
drugs are generally classified based on their actions.
A drug is an agonist if it fully activates receptors, a partial agonist if it partly activates receptors, an inverse agonist if it inactivates receptors (and prevents them
from being activated), or an antagonist if it only prevents receptors from being
activated. For instance, the endogenous neurotransmitter DA is an (full) agonist of
DA receptors, and the antiparkinsonian drug bromocriptine is a partial agonist at
D2 receptors. Antipsychotics like haloperidol and clozapine may be inverse agonists of D2 DA receptors (4,5), whereas L-741,626 is an (neutral) antagonist.
Receptor activation is a thermodynamic process, whereby agonist binding
induces a conformational change in the receptor and converts it from the inactivated state (agonist low-affinity binding state) to the activated state (agonist highaffinity binding state). Typically, neutral antagonist binding is indifferent to the
conformational (affinity) state of its receptor, because it must only occupy the
orthosteric site rather than occupy and then induce a change in it.
However, an
inverse agonist is a special type of antagonist in that when it binds to a receptor in
the activated state it converts it to the inactivated state.
For this reason, inverse
agonists reduce the basal levels of constitutive receptor activity, which corresponds
to the (typically small) proportion of receptors that are in the activated state in the
absence of agonist. Such distinctions in the molecular mechanisms of action of
antipsychotic drugs that act on D2-like dopamine and 5-hydroxytryptamine (5-HT)2-
like serotonin receptors may be critical to understanding their unique therapeutic
profiles (4,6).
Although most drugs bind directly to the orthosteric site of the receptor, other
drugs bind at another (secondary) receptor site, called an allosteric site. Ligands
that bind to the allosteric site are known as allosteric modulators, because they
indirectly modulate the binding of primary ligands to the orthosteric site by
remotely altering the orthosteric-binding site.
The modulation is said to be positive
if the modulator facilitates a primary ligand’s interaction with the primary site or
negative if the modulator attenuates its interaction with the primary site. The extent
to which the allosteric site and the orthosteric site are coupled, or their coopera
Pharmacotherapeutic Principles 33
range of allosteric mechanisms and corresponding allosteric sites exist for modulating the effects of endogenous and therapeutic agents
(7). For example, the
endogenous tripeptide proline–leucine–glycine (PLG) is a positively cooperative
allosteric modulator of agonist binding to D2 DA receptors. Sodium ions are negatively cooperative allosteric modulators of agonist binding to D2 DA receptors, and
zinc ions are neutrally cooperative allosteric modulators of antagonist binding to
D4 DA receptors.
4. PHARMACODYNAMICS OF PHARMACOTHERAPIES
Chemical agents that have therapeutic actions are referred to as drugs.
The term
pharmacodynamics describes what drugs do to the body. Most drugs exert their
actions on the body by interacting with specific sites called receptors. Consequently,
pharmacodynamics deals with the interactions of drugs with their receptor sites.
The most critical drug–receptor properties concern the strength of their attraction
(binding affinity) and functional effects (potency) expressed in units of drug concentration, and the quantity of receptor in the target tissue (receptor density) or the
maximal extent of a receptor’s functional effect (efficacy). The density of receptor
sites is typically expressed as moles receptor per amount of tissue, whereas the
maximal functional effect, which relies in part on receptor density, is usually
expressed as receptor activity per unit amount of tissue.
The functional activity of a
receptor can be measured by a variety of endpoints, ranging from changes in biochemical markers to behaviors.
Two coupled events occur when a drug interacts with its receptor.
First the drug
binds to its receptor, and second it mediates some functional effect that is transduced by the receptor. Although drug binding and receptor activation are coupled,
they are mechanistically distinct molecular processes under the control of unique
receptor microdomains and they can be influenced by different factors.
Consequently, there may not be a direct one to one correspondence linking one process to
the other.
4.1. Determination of Drug Affinity and Maximal Receptor Density:
Ligand–Receptor Binding Interactions
The reversible (noncovalent) binding of a ligand with its receptor is a dynamic
process, which is usually studied in one of two ways. The first way is to measure the
kinetics of binding—the rate of approach to or departure from the equilibrium condition.
The second way is to measure the free energy forces of binding under the equilibrium condition. It is helpful to review the general principles of receptor binding
theory, in order to know what sorts of experiments to perform to extract kinetic and
equilibrium properties of ligand–receptor binding interactions, and additionally, to
know how to interpret the meaning of such properties in the context of drug therapies.
The theoretical construct that allows one to extract the properties that describe
both kinetic and equilibrium types of ligand binding processes and the relationship
between them is referred to as the mass action law.
This law assumes that a ligand
34 Schetz
reversibly binds to a single homogenous population of receptor sites.
Because the
law is restricted to reversible reactions, which are those that can attain an equilibrium condition, the ligand–receptor interaction can be modeled as an equilibrium
reaction. As with all equilibrium reactions, when equilibrium is achieved the rate of
the forward and reverse reactions are equal; at equilibrium, the rate of ligand–
receptor association equals the rate of ligand–receptor dissociation as shown
in Eq. 1.
Rate of association (forward reaction) = Rate of dissociation (reverse reaction) (1)
The rates at equilibrium can be expressed mathematically in terms of reactants
and products as shown in Eqs. 2 and 3.
Rate of association = [LIGAND][RECEPTOR]kon (2)
Rate of dissociation = [LIGAND·RECEPTOR]koff (3)
in which
[LIGAND] is the ligand concentration expressed in units of Molarity (i.e., moles/liter)
[RECEPTOR] is the total receptor concentration expressed in units of Molarity
[LIGAND·RECEPTOR] is the ligand–receptor complex expressed in units of
Molarity
kon is the association rate constant for the binding of a ligand with its receptor
expressed in units of (s-1 M-1)
koff is the dissociation rate constant for the separation of ligand from its receptor
expressed in units of s-1
A mathematical model of receptor occupancy can thus be formulated from the
theoretical expectation at equilibrium by substitution of the equalities in Eqs. 2 and
3 for those in Eq.
1 to yield Eq. 4.
[LIGAND][RECEPTOR] kon = [LIGAND·RECEPTOR] koff (4)
Equation 4 can be rearranged such that all the concentration variables occur on
one side of the equation and all rate constants occur on the other side as shown in
Eq.
5. The ratio of the reactants (ligand and receptor) to products (ligand–receptor
complex) thus equals the ratio of the rate of complex dissociation over the rate of
reactant association.
([LIGAND][RECEPTOR])/[LIGAND·RECEPTOR] = koff/kon = KD (5)
These ratios are also equal to the equilibrium dissociation constant (KD), which
represents the concentration of ligand required to occupy half of the total number of
receptors. The units of KD are Molarity. A series of substitutions and algebraic
manipulations to Eq. 5 (8) puts it in the general form of a rectangular hyperbola
(Eq. 6) to yield Eq. 7.
y = ax/(b+x) (6)
[LIGAND·RECEPTOR] = ([RECEPTOR][LIGAND])/(KD + [LIGAND]) (7)
Pharmacotherapeutic Principles 35
Separation of the dependent and independent variables allows for the graphing
of the data and the extraction of the receptor-binding properties for a ligand that are
the constants in the square hyperbola equation (e.g., a = [RECEPTOR] and b = KD).
In the laboratory, the amount of ligand that is specifically bound to its receptor
([LIGAND·RECEPTOR]) is measured as a function of various ligand concentrations ([LIGAND]), and then the [RECEPTOR] and KD are solved for graphically
(by applying a square hyperbolic math function). A common practice is to introduce a radioactive atom into the ligand (so that it can be detected), incubate various
concentrations of this radiolabeled ligand in a solution containing a fixed amount
of its receptor until equilibrium is reached, and then rapidly separate (so as not to
disrupt the equilibrium condition) the radioligand bound to the receptor ([LIGAND·
RECEPTOR]) from the unbound radioligand in solution ([LIGAND]). The radioactivity of the receptor-bound and (unbound) free radioligand is then quantified in
a radioactivity counter. The resulting data obtained for such a saturation isotherm
type of binding experiment is depicted in Fig. 2.
As can be seen from Eq. 5, the equilibrium dissociation constant can also be
calculated by measuring the kinetic rates of ligand association and dissociation from
its receptor as the binding reaction proceeds toward or away from the equilibrium
condition. This can be accomplished by measuring the amount of radioligand bound
to its receptor as a function of time. Kinetic determinations of KD require two separate experiments (association and dissociation rates) for each KD determination and
provide no information on receptor density. Therefore, they are usually not the
method of choice for determining equilibrium dissociation constant values.
Fig. 2. Example of saturation isotherm data for [3H]mesurguline equilibrium binding to
cloned human serotonin 5-HT2C receptor expressed in COS-7 cells. A saturation isotherm
experiment is conducted by keeping all conditions fixed while varying the concentration of
radioligand. The KD = 0.24 nM and Bmax = 0.5 pmoles/mg protein. (Copyright John A.
Schetz, 2003.)
36 Schetz
Although saturation isotherms have the advantage that they are direct measures
of affinity (KD) and receptor density (Bmax), relatively few radiolabeled ligands are
available, and consequently the binding affinity for most ligands must be determined indirectly. The inhibition constant (Ki
) is an indirect measure of a ligand’s
affinity for its receptor that is numerically equivalent to the equilibrium dissociation constant (Ki
= KD). In contrast to saturation isotherm experiments, in which the
only ligand present is the radioligand, an inhibition type equilibrium binding
experiment examines the ability of a nonisotopic ligand to compete with the
radioligand for binding to the receptor site. The inhibition binding experiment is
performed with a fixed concentration of radioligand and receptor vs increasing concentrations of competing ligand. An inhibition affinity constant for the nonisotopic
ligand is derived from its IC50, which is the concentration of nonisotopic (cold)
ligand required to displace half of the total amount of radioligand bound to the
receptor (Fig. 3). The semilog dose–response curves for competition experiments
take on a sigmoidal appearance. The relative IC50 value extracted from the sigmoidal dose–response curve is then converted, by applying the Cheng-Prusoff transformation (9), to an absolute affinity value (Ki
) that is independent of radioligand
affinity and concentration. The competitive form of the Cheng-Prusoff equation
(Eq. 8) is a measure of receptor occupancy at equilibrium that obeys the law of
Fig. 3. Example of competition binding data for raclopride displacement of [3H]methylspiperone from cloned rat dopamine D2 and D4 receptors expressed in COS-7 cells.
The IC50 is the concentration of competing ligand, which is needed to displace half of the
radioligand occupying the receptors. Note that IC50 values are relative measures that are
dependent on the concentration of radioligand employed in the experiment. In order to convert IC50 values to a concentration-independent equilibrium binding constant (Ki
) a correction factor called the Cheng-Prusoff equation must be applied (9). (Copyright John A.
Schetz, 2003.)
Pharmacotherapeutic Principles 37
mass action, i.e., it assumes that the nonisotopic ligand binds the same receptor in
the same manner as the radioligand—a perfectly competitive inhibition at a single
homogenous population of receptor sites.
Ki
= IC50/(1 + ([RADIOLIGAND]/KD)) (8)
The KD value in Eq. 8 corresponds to the affinity of the radioligand and the
Ki value corresponds to the affinity of the competing ligand.
If the interaction is truly competitive then the linear part of the sigmoidal inhibition curve will have a negative slope equal to unity (pseudo Hill slope = 1).
More
shallow slopes can indicate more than one type of receptor, more than one affinity
state for a single receptor or a negatively cooperative allosteric interaction.
Steeper slopes indicate a positively cooperative allosteric interaction. For example,
agonists can bind different conformational states of the receptor (e.g., high- and
low-affinity states) with different affinities, and in these cases, the apparent slope
will be shallow.
When the slope is different from unity the assumptions of the law
of mass action are violated and a true Ki
value cannot be determined. In practice
many ligand–receptor interactions are not perfectly competitive, which is sometimes indicated by reporting a relative inhibition constant (K0.5). If the difference
between high- and low-affinity states are large enough (e.g., approx 100-fold) the
binding curve will be clearly biphasic, and in these cases, the binding interaction
can be described with a two-state model.
A simple competitive binding model is
usually not appropriate for determining the equilibrium dissociation constants for
allosteric modulators, because they are by definition not acting at the same site on
the receptor as the primary ligand-binding site. Instead Schild-type null pharmacological methods (10) or complex kinetic methods
(11) must be used, in order to
assign an equilibrium dissociation constant that accurately reflects the binding
interaction of the allosteric modulator with its allosteric receptor site.
4.2. Determination of Ligand Potency and Efficacy:
Ligand–Receptor Functional Interactions
Although the description of the binding of a ligand to a receptor provides information about the affinity of the ligand for its receptor, it lacks information about
what sort of response the ligand induces in the receptor once it is bound. In order to
accommodate a response component, additional terms, which describe factors that
affect the functional response can be incorporated into the framework of the receptor occupancy model outlined above. For example, the Ariens equation (12)
expresses receptor activity as a fraction, Afraction, of the maximal activity, Amax, and
equates this activity ratio to the fraction of ligand–receptor complex ([LIGAND·
RECEPTOR]) and the total amount of receptors ([RECEPTOR]) as shown in Eq. 9.
Afraction/Amax = (α[LIGAND·RECEPTOR])/[RECEPTOR] (9)
The term α is a proportionality factor that is an expression of the efficiency of
the coupling of the binding of the ligand with its receptor to its subsequent activation
of a receptor response. This efficiency of coupling term is an acknowledgment that
some agonists, known as partial agonists, promote less than the optimal coupling
38 Schetz
that is required to produce a full response. Consequently, even at maximal receptor
occupancy the maximal response for a partial agonist will be less than for a full
agonist. In other words, the amount of receptor occupancy is not directly proportional to the relative amount of response if the ligand is not a full agonist. The
quantity α thus represents the intrinsic activity of a ligand, which is generally
defined as equal to one for the endogenous agonist, 0 for an antagonist, and in
between 0 and 1 for a partial agonist. Because the endogenous agonist is assumed
to be a full agonist, xenobiotic agonists that produce a greater maximal response
than the endogenous agonist can have an efficacy greater than unity.
Inverse agonists are a special case of negative efficacy. The negative value is
owing to the fact that low levels of receptor can, under normal circumstances,
assume the activated state in the absence of agonist.
This basal agonist-independent
activated state is known as constitutive activity. The concept of negative efficacy
is a result of defining the basal state (agonist-independent activity) as the 0 or
baseline value for agonist-stimulated activity. Because inverse agonists bind to the
activated (high-affinity) state of the unoccupied receptor and convert it to the inactivated (low-affinity) state, they inhibit basal activity and are said to possess negative efficacy. In contrast to an inverse agonist, an antagonist has no effect on basal
activity and because it also is incapable of stimulating the receptor to produce a
functional response it is said to have no efficacy.
From Eq. 9 it can be seen that two important factors controlling the measured
functional activity of receptors in response to ligand binding are receptor density
([RECEPTOR]) and stimulus–response coupling efficiency (intrinsic activity, α).
Like Eq. 7, which describes a saturation binding reaction, Eq. 9 describing the functional response also can be expressed in the form of a rectangular hyperbola (Eq. 6)
to yield Eq. 10.
Afraction = (α Amax[LIGAND])/([LIGAND] + (1/ KD)) (10)
When plotted on a semilogrithmic scale the rectangular hyperbolic function takes
on the form of a sigmoidal curve. Consequently, a plot of the fraction of functional
response (Afraction) vs the logarithmic concentration of drug (log[LIGAND]) can be
fitted with Boltzman’s equation describing sigmoidal functions (Fig. 4). The maximal function response or efficacy for a given ligand is the point in which the functional response reaches a plateau at higher concentrations of ligand (Fig. 4),
although the concentration of ligand that produces half of the maximal response
(Afraction/Amax = 0.5 = EC50) is defined as the potency. Both potency and efficacy
are relative measures whose values rely in part on receptor density. Examples of
the receptor mechanisms underlying the expected functional responses produced
by ligands with different functional activities are depicted in Fig. 4.
The functional response term EC50 and the competitive ligand binding property
IC50 bear some relation to one another, and although it is tempting to try to draw an
analogy between them, there are some important distinctions. Both the EC50 and
the IC50 are terms that correspond to concentrations of ligand that produce a half
maximal measurement (i.e., activity or inhibition of binding). However, the IC50 is
Pharmacotherapeutic Principles 39
a measure of the ability of a competing ligand to inhibit the binding of a radioligand
to its receptor that is both independent of receptor density and directly proportional
to receptor occupancy. The EC50 is a measure of functional effect that is dependent
on receptor density and not necessarily directly proportional to receptor occupancy.
The reason that the functional response is not always directly proportional to receptor occupancy by ligand is that the strength of coupling between binding and
response must be considered. This is not the case for a ligand–receptor-binding
interaction because there is no additional coupling component to consider. This
difference between binding interactions and functional responses is the molecular
explanation regarding why, depending on the ligand’s intrinsic activity and the conditions under which it is tested, a ligand’s affinity value for its receptor may be
different from its potency value.
5. PHARMACOKINETICS OF PHARMACOTHERAPIES
The clinical evaluation of a drug in vivo concerns dose–effect relationships, but
the pharmacodynamic measures of the concentration–effect of drugs, described
above, provide only part of the information. Relating dose to effect requires one to
consider the dose–concentration relationships of a drug and then associate this with
its concentration–effect relationships. A knowledge of pharmacokinetics, which is
what the body does to or with a drug once it is administered, is key to understanding
the relationship between drug dose and attaining a concentration of drug at the
desired target site for an appropriate period of time to produce the intended therapeutic effect.
Because the drug targets for neuropsychiatric disorders are embedded in brain
structures that are not readily accessible, drugs cannot be easily applied directly to
Fig. 4. Examples of responses for ligands with agonist, partial agonist, inverse agonist
and (neutral) antagonist functional properties. The EC50 is the concentration of ligand that
produces a half maximal effect, while the efficacy corresponds to the relative level of maximal effect, which can be denoted as intrinsic activity (α). (Copyright John A. Schetz, 2003.)
40 Schetz
the target tissues. Rather neuropsychiatric drugs must be introduced into the body
at some distal site and then travel to their target sites in the brain. Of great importance to dosing is what happens to a drug once it is administered and en route to its
target site. Although some drugs are applied intravenously in clinical trails,
once their effectiveness is established most drugs are formulated for oral dosing.
The oral administration of drugs is the preferred route of administration for clinical
applications, because it eliminates safety concerns associated with the use of
needles and it facilitates outpatient treatment. Following oral administration and on
its way to its target site,
a drug will encounter various biological barriers, metabolic
tissues, and nontarget tissue deposition sites. The collective effect of these factors
largely determines the amount of intact drug that is free to interact with the intended
receptor target within a given time frame after dosing. Some critical pharmacokinetic parameters to consider for a drug are the time and concentration of its maximal blood levels, its apparent volume of distribution, its rate of clearance and its
half-life.
These parameters depend on the processes of drug absorption, distribution, metabolism, and excretion.
5.1. Absorption of Orally Administered Drugs
and the Time and Amount of Maximal Drug Levels in Blood
For orally administered drugs, absorption begins with the transport of a drug
from the gut to portal blood, continues as the drug passes through the liver, and
ends when the drug reaches systemic circulation. If the drug is metabolized by the
liver or its passage across the gastrointestinal barrier is incomplete,
then the drug
has reduced bioavailability. Bioavailability is defined as the fraction of intact drug
that reaches the systemic circulation relative to the administered dose. With the
exception of replacement strategies, such as L-DOPA treatment for Parkinson’s
disease, most drugs cannot utilize existing active transport mechanisms utilized by
endogenous agents, and consequently, their transport properties are largely determined by passive diffusion across biological barriers. The rate and extent of oral
drug absorption depends strongly on the physiochemical characteristics of the drug,
the formulation state of the drug, and the gastric composition. Because the gut–
blood barrier is comprised of cells with lipid membranes and aqueous interiors, the
passive transport properties of a drug correlates well with partition coefficient measures of its preference for octanol (a lipophilic environment) over water
(a
Pharmacotherapeutic Principles 41
The drug formulation is another factor that can affect absorption of a drug.
For example, oral formulations for the antiparkinsonian drug Sinemet® (L-DOPA
plus carbidopa) can range from a rapidly absorbed liquid to a slowly absorbed capsule and even more slowly absorbed controlled release tablet.
For many therapeutic
applications it is desirable to gradually increase and then maintain steady blood
levels, as rapid rises in blood levels of drugs can desensitize receptor responses or
produce significant adverse side effects
(e.g., nausea in the case of DA receptor
agonists), and large changes in blood levels can result in fluctuating therapeutic
responses. The blood adsorption characteristics of a drug that are usually of most
interest are its maximal blood concentration and the time at which this maximum is
achieved.
5.2. Distribution of Absorbed Drugs and Apparent Volume of Distribution
Distribution is a process involving the exchange of drug in systemic blood with
tissues that it comes in contact with as it travels throughout the body.
Circulating
drugs can either remain soluble in the aqueous blood phase or they can be carried
by blood components. Usually the carrier components in blood are proteins, but in
rare instances, such as for the mood stabilizer sodium valproate, lipids can be the
carrier. In many cases, the drug is not very tightly bound to blood components and
it will prefer to associate with a tissue with which it comes in contact.
Once the
drug has transferred from blood to a tissue it is said to have been distributed or
deposited. In certain cases, a drug may bind so tightly to carrier proteins that it
cannot readily dissociate and interact with other tissues, and the blood proteins then
act as a nontarget tissue deposition sites. Such tightly protein-bound drugs are usually therapeutically inactive in vivo.
Distribution can be a complex process requiring passage across more than one
barrier that separates biological compartments. For example, neuropsychiatric drugs
must cross the bloodbrain barrier (central nervous system compartment) before they
can cross the cellular membrane barriers (cellular compartment) surrounding their
target tissues in the brain. Some drugs can redistribute themselves to the periphery
once deposited in the brain, but this effect is rarely significant for neuropsychiatric
drugs.
More relevant to the pharmacokinetics of neuropsychiatric drugs is the distribution of antipsychotic and antidepressant drugs into lipophilic stores such as
fat.
The reason for this is that antipsychotic and antidepressant drugs tend to be
very lipophilic owing to having a number of aromatic rings. Such antipsychotic or
antidepressant drugs can remain intact when stored in fatty tissues, and they can be
slowly released over time, which can account for their sometimes long washout
period.
On the other hand, the antimania drug lithium is a very water-soluble
elemental ion that distributes in a manner similar to bulk water. Lithium is also
unique among neuropsychiatric agents in that it is not protein bound, and it is primarily transported into cells via passage through voltage-dependent sodium channels.
Once inside cells, lithium is only slowly released, because it does not substitute
for sodium for active transport through the sodium-potassium pump.
42 Schetz
A useful parameter for describing drug distribution is the volume of distribution
(Vd), which is an apparent measure of the accessible space in the body that is available to contain a drug. It can be defined as the ratio of the amount of drug in the
body to the concentration present in the aqueous portion of blood (blood water) as
shown in Eq. 11.
Vd = amount of drug/concentration of drug in blood water
(11)
Vd is only an apparent value, because it often does not relate to the real volume
of the body. Instead, Vd is an operational definition that relates to a volume that
would be required to homogeneously contain drug at the concentration found in
blood. Large volumes of distribution indicate that the amount of drug measured in
the blood is low as a result of distribution of the remaining drug into various tissues.
Drugs that are not highly bound to blood constituents and that readily distribute
into body tissues will have larger volumes of distribution. Note that the Vd values
apply to intravenously administered drugs, unless an orally administered drug is
completely or almost completely bioavailable, otherwise Vd values for an orally
administered drug must be estimated by multiplying Vd by drug bioavailability.
5.3. Termination of Drug Responses
A drug response is terminated by excreting the drug from the body or by metabolically inactivating it. The excretion of a drug from the body depends on its clearance.
Systemic clearance
is a process by which the portion of drug that it is not
metabolized and not protein-bound is removed from systemic circulation by hepatic
excretion into the bile and/or by renal excretion into the urine. Renal excretion is
common for small or polar drugs.
For the majority of neuropsychiatric drugs at the
doses utilized in clinical settings, the clearance is assumed to be a first-order process and is constant.
Another parameter that describes the elimination of drug is the elimination rate
constant (Ke). The constant Ke is the fraction of drug excreted at any instant in time,
and it is a function of clearance and volume of distribution as shown in Eq. 12.
Ke = systemic clearance/Vd (12)
The elimination half-life (t1/2) is the time needed to eliminate half of the drug
from the body. The Ke, or its related clearance and Vd values, can be used to estimate the elimination half-life (t1/2) of a drug as shown in Eq. 13.
Elimination half-life = t1/2 = ((ln(2))(Vd))/systemic clearance = (ln(2))/Ke (13)
The value ln(2) in Eq. 13 is the proportionality constant for the first-order elimination of half of the drug. The elimination half-life value can be utilized to estimate
drug-dosing regimens, the time needed to achieve steady-state levels, and the time
needed to wash out the drug following the last dose. In general, the time needed to
attain steady-state drug levels, or to approximate the drug wash out period is estimated to be greater than five elimination half-lives.
Pharmacotherapeutic Principles 43
The dependence of the elimination half-life on Vd is as a result of the fact that
only drugs that are in systemic circulation and in contact with organs of elimination
(e.g., liver and kidney) can be cleared, although drugs distributed into other tissues
cannot. The clearance of many drugs relies on the rate of blood flow to the organs
of elimination. In these cases, the functional status of the heart, as a result of age,
disease, or drugs that alter cardiac function, can significantly affect clearance,
because blood flow rate is altered.
The functional status of the major organs of
elimination, owing to disease or age, for example, can also affect clearance rates,
and consequently, the elimination half-life of drugs that are cleared by these organs.
For example, impaired renal function, which is common in the elderly populations,
can essentially double the elimination half-life of lithium as it is primarily cleared
by the kidney
(13).
Although clearance is often the predominant factor in the termination of
responses for drugs with low-molecular weights or significant polarity, most drugs
used to treat neuropsychiatric disorders tend to be lipophilic and to have relatively
large molecular volumes.
Thus, the majority of such drugs must undergo biotransformation to more polar metabolites before they can be effectively excreted.
The production of more polar metabolites can occur by enzymatic reactions that
either induce or unmasked polar functional groups (phase I reactions), or that conjugate endogenous polar groups like sugars and polar amino acids (phase II reactions), or both. For example, desimipramine metabolism involves hydroxylation
followed by glucuronidation.
Although many drug metabolites are biologically inactive, some retain activity
or have modified activity. A variety of drugs used to treat neuropsychiatric disorders have active metabolites. For example, desimipramine is an active metabolite
of the tricyclic antidepressant imipramine, and norfluoxetine is an active metabolite of the selective serotonin reuptake inhibitor fluoxetine; in both cases the
metabolites have the same targets as the parent drug.
In other cases, the activity
profile of the metabolites is significantly different from the parent drug. For example,
buproprion selectively blocks the DA transporter over the norepinephrine (NE)
transporter, although one of its hydroxylated metabolites gains significant affinity
for the NE transporter (14,15). In another example, the antipsychotic drug loxapine
is metabolized to the antidepressant amoxapine, which converts it from a D2 DA
receptor-blocking drug to a drug with significantly more norepinephrine transport
blocking activity. Consequently, the metabolism of drugs can either terminate their
actions, by forming inactive metabolites, or when active metabolites are formed,
metabolism can be an underlying reason for their unique pharmacological effects.
6. RECEPTOR RESPONSIVENESS AND TIME OF ONSET
OF THE THERAPEUTIC ACTIONS OF DRUGS
The relationship between drug concentration and functional effect described in
the sections above is for a single challenge of drug at a naïve receptor. Following
prolonged or repeated occupancy, most receptors undergo changes in responsive-
44 Schetz
ness or density that protects them from excessive stimulation or blockade. Such
adaptive responses to the repeated application of drugs can have significant consequences with respect to their actions. For example, attenuated responsiveness may
limit the effective therapeutic use of a drug, it may result in tolerance to side effects,
or it may be the underlying cause for their effectiveness.
Persistent activation as a result of persistent receptor occupancy by agonists leads
to a reduction in receptor responsiveness. In the case of G protein-coupled receptors (GPCR), such as DA receptors, NE receptors, and most serotonin receptors,
attenuated responsiveness is characterized by three types of temporally and mechanistically distinct adaptive processes (16). Persistent receptor stimulation by acutely
administered agonists results in GPCR desensitization followed by internalization.
Receptor desensitization is the result of an uncoupling of the GPCRs from their G
proteins. This uncoupling involves a phosphorylation-dependent (e.g., by kinases)
blocking by cytoplasmic accessory proteins (e.g., arrestins) of intracellular portions of the GPCR that interact with G proteins (e.g., the intracellular loops and
cytoplasmic tail).
Desensitized receptors then undergo internalization whereby
GPCRs are redistributed from plasma membranes to intracellular membranes via
endocytosis. In some case, the internalized receptors are resensitized by dephosphorylation in clathrin-coated vesicles and recycled back to the plasma membrane.
Under conditions of chronic stimulation, internalized GPCRs are not resensitized;
rather, they are downregulated, which leads to a reduction in receptor density owing
to proteolytic degradation. In some cases, chronic stimulation is additionally associated with a reduction in the amount of newly synthesized receptor. Although most
GPCRs display attenuated responsiveness following persistent activation, the rate
and extent of this effect can vary considerably depending on the receptor subtype
and drug pharmacokinetics. In contrast to persistent activation, persistent
blockade of GPCRs can lead to receptor supersensitivity or receptor upregulation.
Neurotransmitter transporters and metabolic enzymes can also display changes in
responsiveness as a result of persistent occupancy, but the details of the molecular
mechanisms are distinct from those described for GPCRs (17,18).
The general expectation is that the onset of drug action will be a function of how
long it takes for a drug to reach its target tissue and then act on its receptor, which
in most cases is rapid. For instance, intravenous bolus injection of the appropriate
dose of phenobarbital into the tail vein of a rat produces sedation in less than 1 minute.
However, the rate of onset of the therapeutic actions of drugs used to treat neuropsychiatric disorders can vary considerably. The anti-attention deficit hyperactivity
disorder effect of psychostimulants, like D-amphetamine and methylphenidate, produce dramatic changes in behavior that closely parallels the expected dose–effect
relationship. In contrast, the onset of action of chronically administered antipsychotic or antidepressant drugs can be much longer, requiring weeks for a full therapeutic effect to be achieved. In these cases, the large disparity between the expected
and actual time course of the therapeutic effect implies that clinical efficacy is not
as a result of acute effects on the target receptor; rather, it is because of chronic
compensatory changes in the target receptor (e.g., up- or downregulation of recep-
Pharmacotherapeutic Principles 45
tor density) or some other receptor system whose function is linked to the target
receptors. For instance, the therapeutic effect of chronic antidepressant treatment
may be as a result of desensitization of presynaptic autoreceptors, such as
somatodendritic serotonin 5-HT1A receptors (19) or terminal serotonin 5-HT1B
receptors (20), and/or downregulation of serotonin transporters (21). The end result
of each of these effects is an increase in the level of synaptic serotonin. A chronic
elevation in synaptic serotonin could be signaling changes in the levels of nuclear
transcription factors, which then regulate the expression of genes related to neurotransmission, and this might also account for the delay between the onset of drug
treatments and their therapeutic effect.
7. THE MEANING OF DRUG SELECTIVITY
When the term “selectivity” is used to describe a drug it can take on a variety of
contextual meanings. Selective effects of drugs can be as a result of differences in
potency, efficacy, or pharmacokinetic accessibility.
However, drug selectivity usually refers to the binding affinity for one receptor (or a subfamily of receptors) over
others.
The most important factors to consider are the relative frame of reference
and the magnitude of the drug selectivity. Although it may be possible to accurately
measure a fivefold difference in the affinity for one drug over another in isolated
tissue fractions or when dealing with cloned receptor systems, for whole tissue in
vitro or in vivo work, in which a large number of potential receptor sites are available, at least a 200-fold difference in affinity is usually required to elicit a truly
selective response.
A selectivity window of this size allows for dosing that will
result in a maximal occupancy of the intended receptor target with little or no occupancy at nontarget receptors. An important caveat with respect to drug selectivity is
that the selectivity of any drug may be difficult to rigorously define,
because it is
not feasible to screen all known related receptor sites and a drug may bind to receptor sites that have yet to be discovered or pharmacologically characterized.
The term “frame of reference” refers to the number of competing targets for a
particular drug. For instance, a compound like NGD 94-1 has an affinity that is
over 500-fold higher for the D4 subtype of DA receptor than for any of the other
DA receptor subtypes (D1, D2, D3, and D5).
It also has over a 500-fold higher affinity for the D4 receptor than for other GPCRs (e.g., serotonin, sigma, and adrenergic
receptors) for which it has been evaluated (22). Thus, within the frame of reference
of receptor sites that were tested, it can be said that NGD 94-1 is a DA D4 receptorselective drug. However, drugs this selective for a particular receptor subtype are
not available for many key receptor systems.
The antipsychotic haloperidol is a
more prototypical example as it binds with high affinity to cloned D2, D3, and D4
receptors (Ki
= 1.2, 4.1, and 1.6 nM, respectively). Because haloperidol has less
than a four-fold lower affinity for the D3 subtype, its in vivo selectivity over the D2
and D4 subtypes is negligible.
However, if the comparison is expanded to include
the entire DA family of receptors, then it can be said that haloperidol is D2-like
selective, as it binds with higher affinity to all members of the D2-like subfamily
46 Schetz
(i.e., D2, D3, D4) than to the D1-subfamily (D1 and D5, Ki
= 63 and 124 nM, respectively). If our frame of reference is among serotonin 5-HT1A, 5-HT2A, and
5-HT2C receptors (Ki
= 2425, 54, and 4475 nM, respectively),
then haloperidol
can be said to be 5-HT2A selective. If we extend our frame of reference to include
both these serotonin receptor subtypes and the entire family of DA receptors, then
haloperidol’s selectivity can be said to be mixed and would thus more accurately be
defined as being 5-HT2A/D2-like receptor selective.
For these reasons, quantitative
in vitro tissue or in vivo studies often must be interpreted with caution, especially if
one neglects to selectively block, with other selective drugs, known sites that are
not of interest.
For instance, low concentrations of the high-affinity serotonin
receptor selective antagonist mianserin and the high-affinity D1-like selective
antagonist SCH23390 could be added to block 5-HT2A and D1-like sites in brain
tissue when using [3H]haloperidol as a radioligand to detect D2-like sites. By analogy, in vivo chemical lesioning with the neurotoxin 6-hydroxydopamine (6-OH) is
usually performed in the present of a norepinephrine transporter inhibitor, like
imipramine, to permit uptake (via catecholamine transporters) into dopaminergic,
but not noradrenergic neurons.
8. TARGETED PHARMACOTHERAPEUTIC MANAGEMENT
OF SELECTED SYMPTOM MODALITIES
Drugs that target dopaminergic and serotonergic, and to a lesser extent
noradrenergic systems, are the ones most often encountered in the pharmacotherapeutic management of the neurological and psychiatric disorders discussed
throughout the following chapters. This may seem odd given the vast array of
unique clinical symptoms observed for the different neuropsychiatric disorders,
but sense can be made of this by realizing that the pharmacotherapies for neuropsychiatric disorders are largely palliative, and usually, they are designed to provide relief for only one of a range of symptom modalities encountered for each
disorder.
For example,
antipsychotic drugs are prescribed for the treatment of
disorders as divergent as autism, Tourette’s syndrome, and schizophrenia, but
their application is designed to alleviate different symptoms associated with each:
aggression and self-injurious behavior for autism, repetitive motor behaviors for
Tourette’s, and psychosis for schizophrenia. Utilization of similar treatments for
different neuropsychiatric symptom modalities is thus possible because of the
key roles that the various dopaminergic pathways play in the modulation of a
range of cognitive and motor functions.
ACKNOWLEDGMENTS
The author thanks Drs. Michael Oglesby, Robert Luedtke, and Anna Ratka for
insightful discussions and helpful comments. This work was supported in part by
grant R01 MH063162-01 awarded to J.A.S.
Pharmacotherapeutic Principles 47
REFERENCES
1. Jones BJ, Blackburn TP. The medical benefit of 5-HT research. Pharmacol Biochem Behav.
2002;71:555–568.
2. Xu Y, Ito A, Arai R. Immunohistochemical localization of monoamine oxidase type B in the taste
bud of the rat. Neurotoxicology. 2004;25:149–154.
3. Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular
biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol
Rev. 1999;51:593–628.
4. Hall DA, Strange PG. Evidence that antipsychotic drugs are inverse agonists at D2 dopamine
receptors. Br J Pharmacol. 1997;121:731–736.
5. Wilson J, Lin H, Fu D, Javitch JA, Strange PG. Mechanisms of inverse agonism of antipsychotic
drugs at the D(2) dopamine receptor: use of a mutant D(2) dopamine receptor that adopts the activated conformation. J Neurochem. 2001;77:493–504.
6. Weiner DM, Burstein ES, Nash N, et al. 5-hydroxytryptamine2A receptor inverse agonists as
antipsychotics. J Pharmacol Exp Ther 2001;299:268–276.
7. Schetz JA. Allosteric modulation of dopamine receptors. Mini-review. Med Chem. 2004; in press.
8. Limbird LL. Identification of receptors using direct radioligand binding techniques. In: Cell Surface Receptors: A short course on Theory and Methods. Martinus Nijhoff Publishing, 1986:51–96.
9. Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (Ki
) and the concentration
of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem
Pharmacol. 1973;22:3099–3108.
10. Ehlert FJ. Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mole Pharmacol. 1988;33:187–194.
11. Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol
Rev. 2002;54:323–374.
12. Ariens EJ. Affinity and intrinsic activity in the theory of competitive inhibition. Arch Int
Pharmacodyn Ther. 1954;99:32–49.
13. Ritschel WA. Pharmacokinetics in the aged. In: Pagliaro LA and Pagliaro AM, eds. Pharmacologic aspects of aging. Mosby, 1983.
14. Ascher JA, Cole JO, Colin, J-N, et al. Bupropion: a review of its mechanism of antidepressant
activity. J Clin Psychiatry. 1995;56:395–401
15. Bondarev ML, Bondareva TS, Young R, Glennon RA. Behavioral and biochemical investigations of bupropion metabolites. Eur J Pharmacol. 2003;474:85–93.
16. Tsao P, Cao T, von Zastrow M. Role of endocytosis in mediating downregulation of G-proteincoupled receptors. Trends Pharmacol Sci. 2001;22:91–96.
17. Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure,
regulation and function. Nat Rev Neurosci. 2003;4:13–25.
18. Kumer SC, Vrana KE. Intricate regulation of tyrosine hydroxylase activity and gene expression.
J. Neurochem. 1996;67:443–462.
19. Hensler JG. Regulation of 5-HT1A receptor function in brain following agonist or antidepressant
administration. Life Sci. 2003;72:1665–1682.
20. Blier P. Pharmacology of rapid-onset antidepressant treatment strategies. J Clin Psychiatry.
2001;62 Suppl 15:12–17.
21. Benmansour S, Owens WA, Cecchi M, Morilak DA, Frazer A. Serotonin clearance in vivo is
altered to a greater extent by antidepressant-induced downregulation of the serotonin transporter
than by acute blockade of this transporter. J Neurosci. 2002;22:6766–6772.
22. Tallman JF, Primus RJ, Brodbeck R, et al. NGD 94-1: identification of a novel, high-affinity
antagonist at the human dopamine D4 receptor. J Pharmacol Exp Ther. 1997;282